



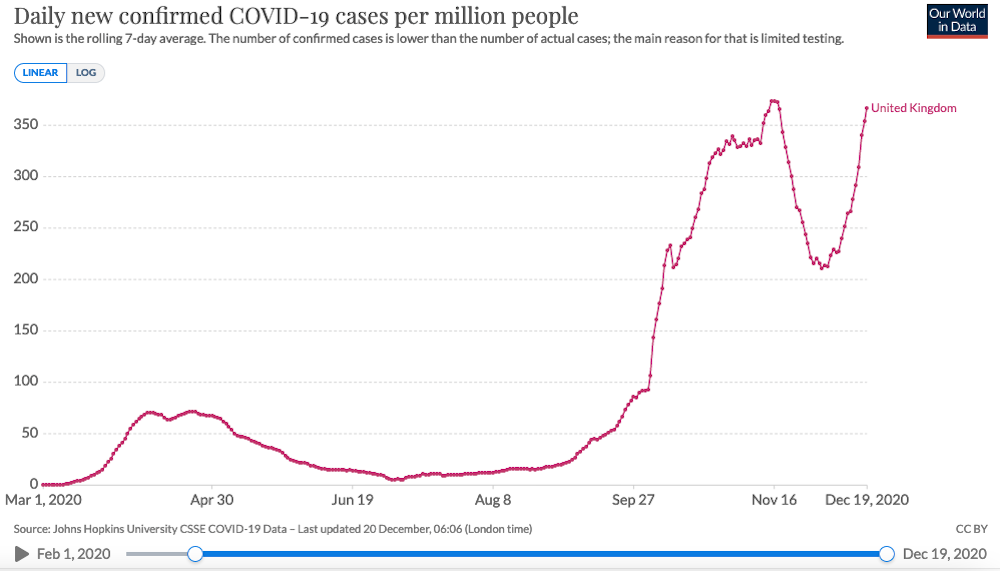
A new variant of SARS-CoV-2, the virus which causes COVID-19, is thought to be driving increased transmission of the disease in parts of the UK. The government has placed some regions including London under new, stricter coronavirus restrictions, known as Tier 4. People in Tier 4 areas will not be able to gather with anyone outside their household for Christmas, while those in the rest of the country can only gather on Christmas Day itself.
Boris Johnson, the prime minister, and his chief scientific advisors said that the new variant could increase transmission of COVID-19 by as much as 70% and increase the R or reproduction number by 0.4.
What’s the significance of this new discovery? The Conversation asked Lucy van Dorp, a microbial genomics researcher and an expert in the evolution of pathogens, some key questions about what we know at this point in time.
What do we know about this new variant?
The new UK variant, known as VUI–202012/01 or lineage B.1.1.7, was first announced by Matt Hancock, the health secretary on December 14. It was subsequently confirmed by Public Health England and the UK’s COVID-19 sequencing consortium. Screening back through databases of SARS-CoV-2 the first sample was taken in the county of Kent on September 20.
The variant carries 14 defining mutations including seven in the spike protein, the protein that mediates entry of the virus into human cells. This is a relatively large number of changes compared to the many variants we have in circulation globally.
To date, genetic profiles – or genomes – of this variant have been largely sequenced and shared from the UK but include some in Denmark and two cases in Australia. There have also been reports of a case in the Netherlands. These countries all have very large genome sequencing efforts and it is very possible that these observations do not reflect the true distribution of this variant of the virus, which could exist undetected elsewhere. We will know more as more genomes are generated and shared.
Thanks to the efforts of data sharing, genomic surveillance and COVID-19 test results in the UK, it seems that this variant is now starting to dominate over existing versions of the virus and that it may be responsible for an increasing proportion of cases in parts of the country, particular in regions where we also have rapidly expanding case numbers.
It is always very difficult to disentangle cause and effect in these cases. For example increases in the appearance of certain mutations can be due to viral lineages carrying them rising in frequency just because they happen to be the ones present in an area where transmission is high, for example due to human activities or choice of interventions.
Though this is still a possibility, there are clearly enough concerning observations so far for this variant to warrant very careful characterization, surveillance and interventions to curb transmission.
Is it more dangerous?
Chris Whitty, the chief medical officer, stated clearly that there was no evidence to date that this variant alters disease severity, either in terms of mortality or the seriousness of the cases of COVID-19 for those infected. Work is underway to confirm this.
How do virus mutations happen?
Mutations are a natural part of virus evolution. In the case of SARS-CoV-2, these mutations may arise due to random errors during virus replication, be induced by antiviral proteins within infected people, or via genetic shuffling – known as recombination. Though signs of recombination are not currently detected in SARS-CoV-2.
Most viral mutations are expected to have no impact. For example, when our team assessed individual mutation replacements in more than 50,000 genomes from the first wave of the pandemic, we detected none that significantly altered viral fitness – the ability of the virus to survive and reproduce.
However, every so often a mutation, or in this case a particular combination of mutations, may strike lucky and offer the virus a new advantage. Viruses carrying these combinations of mutations may then increase in frequency by natural selection given the right epidemiological environment.
Where did the variant come from?
Right now, we don’t know. To date, scientists have not identified any closely related viruses to support the theory that the variant had been introduced from abroad. The patterns of mutations observed are more supportive of an extended period of adaptive evolution most likely in the UK based on current data.
Similar patterns of mutation to these have been observed in the evolution of SARS-CoV-2 in chronically infected patients with weaker immune systems. The current hypothesis is that such a scenario of chronic infection, in a single patient, may have played a role in the origin of this variant. This will continue to be investigated.
How many variations of SARS-CoV-2 have we found?
There are many thousands of lineages of SARS-CoV-2 which differ on average by only a small number of defining mutations. It remains true that SARS-CoV-2 currently in global circulation have little genomic diversity. Subtleties in the mutations carried in different lineages can, however, be very useful for reconstructing patterns of transmission.
As an example, work early in the pandemic used lineage assignments to identify at least a thousand introductions of SARS-CoV-2 into the UK.
Why is this one different?
It is important to note that many of the mutations defining the UK variant have been observed in SARS-CoV-2 before and even sometimes quite early in the pandemic.
Yet the UK variant, or lineage, is defined by an unusual number and combination of mutations. One of these mutations, N501Y, has previously been shown to increase binding of the virus to receptors in our cells. N501Y was first sequenced in a virus in Brazil in April 2020 and is currently associated with a SARS-CoV-2 variant also rising in frequency in South Africa – an independent lineage from B.1.1.7 that is also warranting concern.
The particular deletions identified in the spike protein of B.1.1.7 have appeared in multiple other lineages of the virus at increasing frequency and are also observed in chronic infections where they may alter antigenicity – recognition by immune antibodies. These deletions may also be associated with other mutations in the binding region of the coronavirus spike protein, including those observed in infections among farmed mink and a mutation shown to play a role in the virus’s ability to evade the immune system in humans. B.1.1.7 also harbours a truncated ORF8 gene, with deletions in this region previously associated with decreased disease severity.
The functional effect of these mutations and deletions, particularly when in the combination reported in B.1.1.7, are still to be determined. The high number of mutations and the recent increase in prevalence of this particular variant, together with the biological relevance of some of the mutation candidates, emphasises the need for in-depth study.
What does this mean for the vaccine?
At the moment we don’t know. Though we should be reassured that vaccines stimulate a broad antibody response to the entire spike protein, so it is anticipated that their efficacy will not be significantly hampered by mutations. This is already being tested.
However, there is an increasing body of evidence that other species of seasonal coronaviruses exhibit some ability to escape immunity over longer time periods.
It is therefore conceivable that we may reach a point where we are required to update our COVID-19 vaccines, as we do for influenza, to reflect the variants in circulation at the time. It’s too early to say if this will be the case now, but extensive genome sequencing, data sharing, and standardized reporting of variants will be vital to inform these efforts.![]()
![]()
Lucy van Dorp, Senior Research Fellow, Microbial Genomics, UCL. This article is republished from The Conversation under a Creative Commons license. Read the original article.





